B. Mechanisms of Drug and Nutrient Interactions
There are several mechanisms by which drugs may impact nutrient status and vice versa. The outcome of a drug-nutrient interaction may involve altered disposition of a drug or nutrient (modified absorption, distribution or elimination) – or altered effect of the drug and/or nutrient due to modified actions at the cellular level. Clinical outcomes of a drug-nutrient interaction can range from compromised nutrition status to altered therapeutic response.[1]
This section will describe opportunities for drug and nutrient interactions that may occur during processes of drug ingestion or administration, absorption, distribution, metabolism and excretion. Section C will define specific examples.
| Ingestion/ Administration |
|
| Absorption |
|
| Distribution |
|
| Metabolism |
|
| Excretion/ Elimination |
|
Ingestion/Administration
The mode of a drug’s administration will directly impact its pharmacokinetics. A drug may enter the body in a variety of ways: as an oral liquid, pill or capsule; inhaled as a vapor or aerosol; absorbed through the skin; injected into muscle, subcutaneous tissue, spinal fluid, or directly into the bloodstream.[3] Drug ingestion can cause changes in appetite, food intake and absorption (e.g., taste disorder, nausea, vomiting, diarrhea), thereby indirectly altering nutrition status.[4]
Absorption
For a drug to “work”, it must be absorbed into the circulation to reach a site of action. Most drugs are absorbed by passive diffusion across a biological barrier (such as a cell membrane) and into the circulation.[5] The rate of absorption is proportional to the drug concentration gradient across the barrier, and the surface area available for absorption at that site; this is known as Fick’s law (i.e. the higher the concentration, the higher concentration of drug absorbed).[6] Drugs may be absorbed passively through cells either by lipid diffusion or by aqueous diffusion, depending on their solubility. Lipid diffusion is a process by which the drug dissolves in the lipid components of cellular membranes, this process is facilitated by a high degree of lipid solubility of a drug. Aqueous diffusion occurs by passage through aqueous pores in cell membranes. Aqueous diffusion is restricted to drugs that have low molecular weights, many drugs are too large to be absorbed by this process.[7]
A few drugs require energy or a specialized carrier to be absorbed. Drugs that are absorbed by active transport require a specialized “carrier” molecule and a form of energy, provided by hydrolysis of the terminal high-energy phosphate bond of adenosine triphosphate (APT).[8] In the previous paragraph the process of passive diffusion was described. Passive diffusion does not require energy; drug molecules move “down” a concentration gradient (i.e. from a higher to lower concentration). However, active transport can transfer drugs against a concentration gradient. Facilitated diffusion also requires a carrier molecule; however, no energy is required.[9]
Oral administration (PO). When a drug is swallowed, it must be absorbed from the gastrointestinal (GI) tract into the portal circulation. Many drugs are well absorbed from the GI tract.[10] However, there are a number of drug classes that cannot be administered orally. Biologics (i.e. antibodies) are usually too large, and protein drugs are destroyed in the digestive process. The rate and range of drug absorption by the GI tract is a function of physiochemical properties of the drug (water or lipid solubility), its formulation (tablet, capsule, liquid, time-release) and physiological environment (pH of the stomach).[11] With oral administration, foods and nutrients can impact the absorption of a drug by binding to it or by changing its environment (e.g., pH of the stomach). Food ingestion releases digestive enzymes that can inactivate certain drugs.[12] Oral administration is the route by which most drug and nutrient interactions occur. Interestingly, the bioavailability of some drugs is increased with food, and for others, decreased. The variance is due to differences in drug solubility, molecular size and other chemical characteristics.[13]
Certain classes of antibiotics (e.g. tetracyclines and fluoroquinolones) bind divalent and trivalent cations to form a nonabsorbable complex, so taking them at the same time that you take certain dairy products, or iron/calcium supplements should be avoided.[14]
The intravenous (IV) route of drug administration delivers the drug directly into the bloodstream, to the heart and on to the general circulation.[15] This route bypasses challenges facing absorption from the GI tract and first pass metabolism and allows for quick adjustment of dose to effect. First pass metabolism (or the first pass effect) refers to the rapid uptake and metabolism of a drug by the liver, before it reaches the systemic circulation. First pass metabolism reduces the bioavailability of a drug to less than 100% when processed by the liver before reaching circulation.[16]
Other modes of drug administration include intramuscular, subcutaneous, inhalation, transdermal and topical routes. Intramuscular (IM) drug injection/ administration results in rapid drug absorption because of the high vascularity of muscles. Subcutaneous (SC) administration involves drug delivery into tissue beneath the skin and its subsequent intro into the blood perfusing the tissue.[17] Absorption following SC administration is generally rapid. Inhalation is a rapid route for drug absorption because of the large surface area and high vascularity of the lungs.[18]
Distribution
Drugs are distributed to target organs, tissues and cells via the circulation by diffusing into interstitial fluid and cells from the bloodstream. The extent of any given drug’s distribution is dependent on these factors: lipid solubility and molecular size of the drug, organ blood flow/perfusion and plasma protein binding.[19] Patients with a higher BMI (body mass index) could require differential dosing and it may be expected based on body composition that lipophilic drugs have a larger volume of distribution in obese patients.[20] While beyond the scope of this course, it is worth mentioning that a notable restriction to drug distribution is the blood brain barrier (also called BBB). The blood brain barrier is a network of blood vessels and tightly packed cells which functions to keep out bacteria, some drugs and other harmful substances. It allows water, oxygen, carbon dioxide, and general anesthetics to pass into the brain.[21]
Metabolism
Metabolism gradually decreases plasma drug concentration over time. Metabolism describes the chemical reactions that convert drugs into compounds which are easier for the body to eliminate and excrete.[22] Drug metabolism is also known as biotransformation and is a key step toward eliminating or disposing of a drug. Drug metabolism could be viewed as a detoxification process and ultimately makes a drug more water soluble so that excretion processes (e.g. biliary, renal) can finish disposing of it.[23]
It should be noted that all organisms are constantly (and unavoidable) exposed to xenobiotics including man-made and natural chemicals such as drugs, plant alkaloids, microorganism toxins, pollutants, pesticides, and other industrial chemicals.[24] Biotransformation – or metabolism – of these compounds are generally subdivided into phase I and phase II metabolic reactions. These may or may not occur in “sequential” order. In many cases, phase 1 enzymatic reactions can create or unmask a chemical group that is required for a phase 2 reaction. However, drugs may bypass phase 1 and go directly to phase 2 metabolism. Some phase 1 metabolites are pharmacologically active; however, most phase 2 metabolites are inactive.[25]
Drug excretion is the removal of drugs from body fluids and occurs primarily in the urine. Other routes of excretion from the body include in bile, sweat, saliva, tears, feces, breast milk, and exhaled air.[26] Drugs are handled by the kidneys in the same manner as are endogenous substances, undergoing processes of glomerular filtration, active tubular secretion, and passive tubular reabsorption.[27]
Phase 1 metabolism, or biotransformation, involves oxidative, reductive and hydrolytic reactions.[28] Microsomal cytochrome P450 (CYP) enzymes found in the endoplasmic reticulum of liver and intestinal mucosa cells are critical to the metabolism of endogenous and exogenous chemicals (drugs). These enzymes are also important for many diet-drug and drug-drug interactions. Many drugs can either inhibit or induce one or more of these enzymes and can influence the clearance of other drugs.[29] CYP enzymes have been extensively studied, cloned, and their role in drug metabolism is well established. CYP enzymes and isozymes (two or more enzymes with identical functions but different structures) are divided into three families: CYP1, CYP2 and CYP3. Each CYP enzyme is denoted by a numeral designating the family (e.g., CYP1), a letter indicating the subfamily (e.g., CYP1A), and a number representing the individual enzyme (e.g., CYP1A2) (1). Up to fifty percent of prescription drugs are metabolized by CYP3A4.
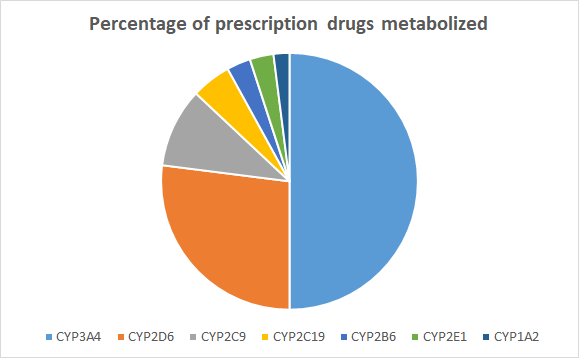
Figure 1.2. Relative contribution of enzymes involved in drug metabolism[30]
Therefore, inhibition of CYP enzymes or isozymes prevents their action on a drug compound, causing its plasma concentration to rise.[31]
Phase 2 metabolism, or biotransformation, involves conjugation reactions that make partially metabolized drug compounds more water soluble, thereby promoting excretion.[32] On the other hand, these conjugations also can play an essential role in the toxicity of many chemicals due to the metabolic formation of toxic metabolites such as reactive electrophiles. Gene polymorphism of biotransformation enzymes may often play a role in various pathophysiological processes. Phase II metabolism, or conjugation reactions, attach small hydrophilic molecules (i.e., acetate, glucuronic acid, sulfate, or glycine) to create more water-soluble compounds, allowing for eventual elimination.[33]
Glucuronidation, carried out by UDP-glucuronosyltransferases (UGTs) belong among key enzymes of metabolism for various exogenous and endogenous compounds.[34] Conjugation reactions catalyzed by the superfamily of these enzymes serve as the most important detox pathway for a broad spectrum of drug, dietary chemicals, carcinogens and environmental chemicals. Glucuronide formation is the most common conjugation reaction (40-70% of clinically used drugs) and uses glucuronosyltransferases to conjugate a glucuronate molecule with the parent drug molecule.[35]
Sulfoconjugation (or sulfonation) catalyze the conjugation of several drugs and endogenous compounds. The enzyme family mediating sulfoconjugation reactions are called sulfotransferases (SULTs). Acetaminophen as well as dopamine undergo sulfonation.[36]
Acetylation is accomplished by N-acetyltransferase enzymes that use acetyl coenzyme A (acetyl CoA) as a source of the acetate group.[37] Compared to sulfonations and glucuronidations, acetylations are modest in terms of the number and variety of substrates. Acetylation reactions are characterized by the transfer of an acetyl moiety, the donor generally being acetyl coenzyme A, and the accepting chemical group is a primary amino function.[38] Drugs and other foreign compounds that are acetylated in intact animals are either aromatic amines or hydrazines, which are converted to aromatic amides and aromatic hydrazides.[39]
Conjugation reactions are a key mechanism for maintaining homeostasis during exposure to various xenobiotics, such as drugs, industrial chemicals, or food procarcinogens.[40] In humans and many other mammals, the liver is a major site of expression of xenobiotic-metabolizing enzymes, but extra-hepatically localized enzymes also are of great importance. In the intestines, several drug metabolizing enzymes decrease the bioavailability of orally administered drugs or activate environmental carcinogens.[41]
Excretion
Excretion, or elimination, refers to the removal of drugs from the body. The biological effects of exogenous substances (i.e., synthetic or natural compounds) are terminated by the processes of metabolism and excretion. Multiple factors affect the rate and extent of excretion, and accumulation occurs if the rate of absorption and distribution of a drug or a nutrient exceeds the rate of excretion. The kidneys are the major route for excretion of many drugs; body fluids and occurs primarily in the urine. Other routes of excretion from the body include in bile, sweat, saliva, tears, feces, breast milk and exhaled air. Drugs are handled in the kidneys in the same manner as are endogenous substances, undergoing processes of glomerular filtration, active tubular secretion, and passive tubular reabsorption.[42]
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p.; Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p.; Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p.; Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p.; Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p.; Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p.; Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p.; Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p.; Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p.; Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p. ↵
- Brenner GM, Stevens CW. Pharmacology. Fifth edition. ed. 1 online resource (579 pages) p.; Boullata JI, Armenti VT, SpringerLink (Online service). Handbook of drug-nutrient interactions. New York, NY: Humana Press; 2010. ↵
Also known as the first-pass effect; this refers to the rapid uptake and metabolism of a drug agent by the liver before it reaches systemic circulation
The chemical reactions that convert drugs into compounds that are easier for the body to eliminate and excrete; also known as biotransformation
The alteration of a substance (such as a drug), within the body.
Refers to the action of the cytochrome P450 (CYP) oxidative enzyme system; CYP monooxygenases are phase I enzymes.
Refers to conjugation reactions that involve the addition of intracellular polar groups (glucuronate, glutathione, sulfate, and glycine) to the foreign molecules (partially metabolized drug compounds) and function to protect humans against chemical insult by facilitating excretion
